一、前言
二、「藥證」與「FDA」三、505(b)(1),俗稱新藥
四、505(b)(2),俗稱改良新藥
五、505(j),俗稱學名藥
六、市場上的實際案例
一、前言
相信大家剛開始研究生技股的時候,一定會常看到「某某公司研發XX新藥」、「某藥即將進行第X期解盲」、「某公司改良新藥,市場價值高達XX億元」這些字句。
阿到底這些是啥咪東西?學名藥?新藥?品牌藥?改良新藥?
今天用淺顯易懂的方式,讓大家能夠快速的了解相關的差異
以後在看報導的時候,就能夠釐清相關資訊避免頭腦混亂了
二、「藥證」與「FDA」
- 藥證(Marketing Authorization):一國政府核發的「藥品上市許可證」,拿到才能合法生產、進口、賣給病人。每個國家自己管:臺灣是 TFDA、美國是 FDA,歐盟為EMA。
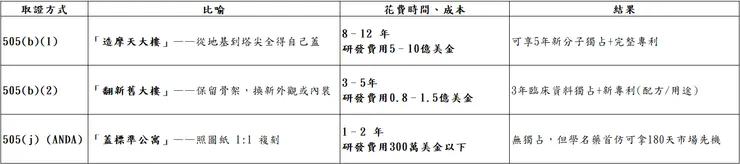
- 美國 FDA(Food & Drug Administration):美國是全球最大的藥品市場!故FDA也是全球最具影響力的藥品審查機關;FDA 透過 505 條款 管理新藥與學名藥申請,制訂了三條「拿藥證」的高速公路:505(b)(1)、505(b)(2) 與 505(j)。
換句話說,想要把藥在美國上市,就必須通過FDA的審核通過拿取藥證,常見方式三種:
三、505(b)(1),俗稱新藥
1、藥物發現
----找出想要應對的疾病,從千萬顆化合物中找出「幾顆」有機會的先導化合物(俗稱:種子Seed),這部分未來有AI參與的機會!
2、前臨床(GLP)
----先用動物、細胞來實驗,證明種子的安全、有效和可製造性
3、提交 IND (Investigational New Drug Application)
----向 FDA 申請「讓新藥進人體試驗」的通行證
4、臨床1期 - 2期 - 3期
----1期:先保命——別讓人「第一口就出事」。
----2期:別浪費時間——確認劑量和初步成效,避免把無效藥拉去燒大錢。
----3期:做最嚴謹比較——用足夠病人數量、雙盲隨機,證明真正價值。

5、CMC (Chemistry Manufacturing Controls)最終化與長期穩定度
----這顆藥怎麼做、做出來長什麼樣子、放著會不會壞?
----FDA必須確定:每一批藥的品質都跟臨床試驗用藥一模一樣或更好,患者才安全。
6、撰寫並提交 NDA(New Drug Application) :505(b)(1)
7、FDA 審查(10 月標準 / 6 月優先)
----Day 0:FDA 收到 NDA/505(b)(1) 或 505(b)(2) 全檔 → 先做 60 天「完整性檢查」
----Day 60:檔案被「Filed」後,正式進入 PDUFA 審查時鐘 (Review Clock)。
----接下來會進入標準審查和優先審查:
(1)標準審查:10個月,針對大多數新藥和改良型新藥
(2)優先審查:6個月,嚴重或罕見疾病且無有效療法、可顯著改善治療安全or療效
8、結果與常見缺失

最後全部通過,才能拿到藥證,準備上市。
四、505(b)(2),俗稱改良新藥
1、找參考藥 (RLD)
----需要選定一個已經被 FDA 核准、且市場上正在賣的「原廠藥」來當比較對象──這個「原廠藥」就叫 RLD。
2、前臨床補件
----確認這個新的藥沒有問題:
(1)小動物毒性追加試驗(如果劑型或劑量變動)
(2)細胞/受控試驗,證明新配方不會產生新毒素
3、IND 提交及溝通
----向 FDA 送開發計畫草稿,並收集 FDA 回覆:需要做哪些 PK / BE 或小型功效試驗
4、Pilot BE&Pivotal BE ± Bridging 試驗
(1)Pilot BE:
試水溫、找配方方向
先用少量志願者測「新配方」 和「原廠藥」的血中濃度,如果計算出來落在 90 %的信賴區間 —or 至少大部分都落在 80 %~125 % 這個安全框,就表示新配方的體內暴露「很可能」已跟原廠藥差不多,接下來做大規模的 Pivotal BE 才有成功的把握。
(2)Pivotal BE:
正式交卷用;核准與否看它
需要做AUC和Cmax兩種試驗:
AUC:新藥吃下去後,血中藥物濃度 vs. 時間 畫出來的面積─代表病人「總共吸收到多少藥」;如果 AUC 差太多,療效或毒性就會不同。
Cmax:最高血中濃度的「高點」;一口氣衝太高容易副作用;太低則效果不夠。
必須讓 AUC、Cmax 的 90 % 信賴區間完整落在 80-125 %。
(3)Bridging 試驗:
新配方差距太大時,FDA可能會請你做一些功效試驗,以證明藥效跟舊藥一致
5、CMC (Chemistry Manufacturing Controls)
同前面所述,製造的品質和穩定度。
6、提交 NDA(505(b)(2)
7、FDA 審查
同前面所述,看BE 統計、CMC 長穩、查廠缺陷。
五、505(j),俗稱學名藥
1、選定 RLD
在 FDA 官方「Orange Book」找出要複製的原廠藥。像影印一份文件前,要先決定用哪一張當原稿。
2、製程與品質對標
把自己的配方、雜質、溶出速度,調到跟原廠藥幾乎一模一樣。目標是讓病人吃了分不出差別,實驗室也測不出大落差。
3、單/雙交叉 BE 試驗
找 24–60 位志願者,一半先吃原廠藥、再吃你的藥;另一半相反。抽血比濃度,如果 90 % 信賴區間落在「80–125 %」安全框,就算兩藥“體內一樣”。
4、CMC 與穩定度
把製造流程寫成SOP,證明每一批都合格。做 ≥12 個月「擺在架上」測試,證明放得久也不變質。
5、提交 ANDA (簡易新藥上市程序)
對現有已獲許可藥物或已批准藥物進行美國仿製藥批准的申請。
6、FDA 技術審查(10 月標準、8月優先)
(1)標準 ANDA:10 個月內審完並「行動」(核准或補件函)。
(2)優先 ANDA(Priority ANDA,限缺藥或競爭不足等情況):8 個月 內審完──比標準快 2 個月,而不是 6 個月。
六、市場上的實際案例:
(一)台康生技,113/12/10的重訊:
1、美國食品藥物管理局(FDA)對本公司專屬授權夥伴Sandoz生物相似藥EG12014凍晶注射劑150毫克藥證申請,提出完全回覆信函(CRL)。

2、這裡就是在申請藥證時,FDA在工廠現地查核時,發現冷凍乾燥填充廠房有生產製造及設施的一些缺失,所以CRL表示:「目前還不能核准,要先修正缺失」。
3、而只要是「製造缺失」且無需重做臨床,常見審查時鐘為 6 個月內 重新審完;若複查排程擁擠,最慢約 9–12 個月。
4、故台康生需於10 天內回覆 FDA,提交初步「CAPA 計畫」。30–90 天內完成整改並提供佐證(SOP、 執行記錄、驗證報告)。若 CAPA 審核通過,FDA 才會核發 BLA/ANDA 核准函 (Approval Letter)。
(二)漢達,113/8/8的重訊:
1、代子公司Handa Oncology, LLC公告505(b)(2)新藥 Vysentri HND-033於美國FDA之查驗登記審查進度

2、這顆藥屬於505(b)2的改良新藥,其中低脂餐試驗的 1 個指標超出 80–125 % 的 BE 標準,故公司 8 月 8 日立即開會,討論將此 PK 結果 與其他現有 PK/PD+文獻一起統計分析,並擬先與 FDA 交換意見,再決定是否補做試驗或修改劑量。
3、由於此前,漢達就已經在112年4月27日接獲FDA對HND-033的完全回應信函CRL,FDA要求重新進行試驗。且漢達於113年4月3日向US FDA申請CRL補件期限展延,並於4月11日取得FDA核准同意展延補件期限6個月,故等於10月底漢達就得交出資料,才有辦法取得藥證,然8月7日又發現上述問題。
4、後續漢達在與FDA溝通並評估CRL補件方向後。初步評估後續準備作業可能超過CRL補件回覆之期限(美國時間113年10月27日),故Handa Oncology, LLC於113年10月17日(美國時間113年10月16日)向FDA申請展延CRL補件期限12個月,最終也獲准,讓漢達有了1年的時間來處理回復CRL的事宜。
(三)全福生技,112/12/21的重訊:
1、公告本公司乾眼症新藥BRM421第一次三期臨床試驗數據解盲之分析結果

2、這是全福生技在開發乾眼症新藥時,臨床試驗的第三期公布結果,我並沒有截取全部的實驗說明,但簡單來說,新藥在臨床實驗中並未達到統計顯著意義,也就是俗稱的解「盲」失敗,這個「盲」就是雙盲,受試者和研究人員都不清楚誰是對照組和實驗組。
※最後不免俗要告誡大家:單純新藥、改良新藥研發,夢想很大,風險也很高,投資前一定要審慎思考!


























